

Allgemeine Informationen
Auf diesen Seiten möchten wir Sie über neue Möglichkeiten der Hämophilie-Therapie informieren. In den letzten Jahren hat sich in diesem Bereich unglaublich viel getan, was einerseits zu Euphorie, andererseits aber auch zu Verunsicherungen geführt hat.
Wir möchten Ihnen hierzu möglichst umfassende und verständliche Informationen bieten.
Melden Sie sich gerne bei uns, wenn Sie Fragen haben. Bei Fragen zu Ihrer persönlichen Therapie wenden Sie sich bitte an Ihren behandelnden Arzt.
Weitere Informationen über neue Behandlungsmöglichkeiten finden Sie auch in den "Novel Treatment Products Newsletter" (auf Englisch), die vom Europäischen Hämophiliekonsortium (EHC) erstellt wurden.
Faktorenkonzentrate mit längerer Halbwertszeit
Wiederkehrende Gelenkblutungen führen zur Schädigung der Gelenke und zur Entwicklung der hämophilen Arthropathie. Zur Vermeidung von Blutungen und den sich daraus entwickelnden Folgeschäden ist die Prophylaxe mit Faktorenkonzentraten bei schwerer, zum Teil auch bei mittelschwerer Hämophilie A und B Therapiestandard. In zahlreichen Studien konnte gezeigt werden, dass durch eine frühzeitig begonnene Prophylaxe (primäre Prophylaxe) Gelenkschäden weitestgehend verhindert werden konnten. Kann die Prophylaxe erst bei bereits manifesten Gelenkschäden begonnen werden, ist das Ziel, die Blutungsfrequenz zu verringern und die Lebensqualität der Patienten zu verbessern.
In den letzten Jahrzehnten haben sich sowohl plasmatische als auch rekombinante (gentechnologisch) hergestellte Faktorenkonzentrate weiterentwickelt, d.h. sie sind sicherer betreffend einer möglichen Übertragung von Viren und auch anwenderfreundlicher geworden (geringeres Volumen, größere Faktorenmenge pro Fläschchen). Dennoch bedeutet die regelmäßige, mehrfach wöchentliche venöse Punktion eine große Belastung für viele Patienten. Einige Patienten bluten zudem auch noch trotz intensiver Prophylaxe und benötigen höhere Faktorenspiegel im Blut. Neuere Studien zeigen, dass ein Faktor VIII- oder IX-Talspiegel von 1% nicht ausreichend zu sein scheint. Dies trifft auch für die Prävention sogenannter Mikroblutungen (kleinste Blutungen in der Gelenkschleimhaut) zu, so dass heutzutage eher höhere Talspiegel (>3%) bei vielen Patienten angestrebt werden.
In den letzten Jahren wurden Faktorenkonzentrate entwickelt, bei denen durch verschiedene Technologien die Halbwertszeit verlängert werden konnte, sogenannte EHL (Extended-Half-Life, englisch Halbwertszeit)-Faktorenkonzentrate. Eine Halbwertszeitverlängerung ist Voraussetzung, um dem Patienten längere Applikationsintervalle und/oder höhere Faktortalspiegel im Rahmen der Prophylaxe bei Hämophilie zu ermöglichen.
Was bedeutet Halbwertszeit?
Die biologische Halbwertszeit ist die Zeit, die der Körper braucht, um die zugeführte Menge eines Stoffes (hier Faktorenkonzentrat) auf die Hälfte wieder abzubauen. Dieser Abbau ist ein natürlicher Vorgang.
Die Faktor-VIII-Halbwertszeit beträgt 8-12 Stunden und ist vom Alter und der Blutgruppe abhängig. Die Faktor IX-Halbwertszeit beträgt ca. 18 Stunden. Hierdurch ergeben sich die klassischen Prophylaxeregime mit FVIII- bzw. FIX-Konzentraten mit Standardhalbwertszeit (plasmatische Faktor-VIII- bzw. IX-Konzentrate, klassische rekombinante Faktor IX- oder FVIII-Konzentrate), die üblicherweise 3x wöchentlich bzw. alle 2 Tage bei der Hämophilie A und 2 x wöchentlich oder alle 3 Tage bei der Hämophilie B durchgeführt werden.
Die Halbwertszeit kann individuell sehr verschieden sein und hat damit Einfluss, wie oft und wie viel sich ein Patient zur Prophylaxe spritzen muss. Das Prophylaxeregime wird natürlich nicht alleine durch die Halbwertszeit bestimmt. Weitere wichtige Faktoren wie Gelenkstatus, Durchbruchsblutungen, körperliche Aktivität, Venenstatus und persönliche Erwartungen spielen bei der individuell gestalteten Prophylaxe ebenfalls eine große Rolle.
Welche Faktorenkonzentrate mit längerer Halbwertszeit gibt es und wie wird deren Halbwertszeit verlängert?
FVIII-Konzentrate mit längerer Halbwertszeit
Es gibt aktuell 5 zugelassene FVIII-Konzentrate mit einer längeren Halbwertszeit, bei denen durch verschiedene Technologien eine Verlängerung der Halbwertszeit auf das 1,2-1,9-Fache erzielt werden konnte (s. Tabelle 1). Weitere Konzentrate befinden sich noch in der Entwicklung (klinische Studien).
Bei Elocta® (Sobi) handelt es sich um ein sog. Fusionsprotein. Hier wird ein rekombinantes FVIII-Molekül mit einem rekombinant hergestellten Teil eines Immunglobulins G (IgG), dem Fc-Teil, verschmolzen. Da das Fc-Teil des IgG eine wesentlich längere Halbwertszeit als FVIII hat, wird die Halbwertszeit von FVIII verlängert. FVIII und auch das Fc-Teil werden vom Körper problemlos abgebaut.
Bei Adynovi® (Shire), Jivi® (Bayer) und Esperoct® (Novo Nordisk) erfolgt die Halbwertszeitverlängerung durch eine PEGylierung von rFVIII. PEG (Polyethylenglycol)-Moleküle gibt es in unterschiedlichen Größen.
Durch die PEGylierung wird das FVIII-Molekül vor vorzeitigem Abbau geschützt. PEG wird im Körper nicht abgebaut und wird unverändert über die Niere oder Leber ausgeschieden. Im Rahmen der Anwendung mit Gerinnungskonzentraten wurde in Studien keine Akkumulation über die Zeit beobachtet, Selten kann es zu einer Unverträglichkeit von PEG kommen (PEG-Antikörper). Dieses Phänomen wird auch bei nicht an Hämophilie erkrankten Menschen beobachtet, da PEGylierung seit vielen Jahren zur Stabilisierung und Halbwertszeitverlängerung von Medikamenten, in der Kosmetik- und auch Lebensmittelindustrie eingesetzt wird. PEGylierte FVIII-Konzentrate sind innerhalb der Europäischen Union für Patienten über 12 Jahre zugelassen.
Die gerinnungsphysiologische Funktion von Faktor VIII wird durch die PEGylierung nicht beeinträchtigt.
Bei Adynovi® binden 20kDa-PEG-Moleküle kontrolliert an FVIII und verlängern so die Halbwertszeit von rFVIII.
Auch bei Jivi® (Bayer) erfolgt die Halbwertszeitverlängerung durch eine PEGylierung, in diesem Fall mit einem größeren, verzweigten 60 kDa-PEG-Molekül.
Bei Esperoct® (Novo Nordisk) erfolgt eine ortsspezifische Bindung von 40kDa-PEG-Molekülen an einem Aktivierungspeptid (Brückenprotein).
Bei Afstyla® (CSL Behring) verändert sich durch eine Veränderung der Gensequenz des FVIII die Bindung zwischen der schweren und leichten Kette des FVIII-Moleküls zum Einzelkettenmolekül. Dieses Molekül hat eine verstärkte Bindung an im Körper natürlich vorkommenden von-Willebrand-Faktor (vWF) und wird dadurch besser stabilisiert.
Bei den 5 zugelassenen Konzentraten werden also unterschiedliche Technologien zur unterschiedlichen Verlängerung der Halbwertszeit angewandt.
Mit allen genannten Konzentraten konnten die Injektionsintervalle im Rahmen einer Prophylaxe verlängert werden, üblicherweise auf 2 x wöchentlich stattfindende Gaben, bei einzelnen Präparaten und Patienten auch auf bis zu 1 x wöchentliche Gaben. Alternativ kann auch der Talspiegel erhöht werden, wenn die übliche Injektionsfrequenz beibehalten wird. Alle Präparate sind gut verträglich und wirken außerhalb der Prophylaxe auch bei der Behandlung von Blutungen oder bei operativen Eingriffen ausgezeichnet.
FIX-Konzentrate mit längerer Halbwertszeit
Bei den 3 zugelassenen FIX-Konzentraten mit verlängerter Halbwertszeit (englisch: Extended Half Life, EHL) konnte ebenfalls durch verschiedene Technologien eine Halbwertszeitverlängerung erzielt werden. Der Effekt ist jedoch im Vergleich zu den FVIII-Konzentraten mit längerer Halbwertszeit wesentlich stärker, denn die Halbwertszeit ist bei den FIX-EHL-Konzentraten um das 3-5fache länger. Dies bietet einen großen Spielraum, was die Erhöhung des FIX-Talspiegels und die Senkung der Injektionsfrequenz bei der Prophylaxe und auch bei der Behandlung von Blutungen/operativen Eingriffen betrifft. Üblicherweise erfolgen die prophylaktischen Gaben nur noch 1mal wöchentlich, aber auch seltenere Gaben sind möglich.
Bei Alprolix® (Sobi) handelt es sich um ein sog. Fusionsprotein. Hier wird ein rekombinantes FIX-Molekül mit einem rekombinant hergestellten Teil eines Immunglobulins G (IgG), dem Fc-Teil, verschmolzen. Da das Fc-Teil des IgG eine wesentlich längere Halbwertszeit als FIX hat, wird die Halbwertszeit von FIX verlängert. FIX und auch das Fc-Teil werden vom Körper problemlos abgebaut.
Bei Idelvion® (CSL Behring) handelt es sich ebenfalls um ein sog. Fusionsprotein. Hier wird ein rekombinantes FIX-Molekül mit rekombinant hergestelltem Albumin verschmolzen. Albumin ist ein im Körper natürlich vorkommendes Eiweiß und hat eine deutlich längere Halbwertszeit als FIX, wodurch die Halbwertszeit von FIX verlängert wird. FIX und auch das rekombinante Albumin werden vom Körper problemlos abgebaut.
Bei Refixia® (NovoNordisk) erfolgt die Halbwertszeitverlängerung durch eine GlycoPEGylierung von rFIX. Es erfolgt eine ortsspezifische Bindung von 40kDa-PEG-Molekülen an einem Aktivierungspeptid (Brückenprotein). Durch die PEGylierung wird das FIX-Molekül vor vorzeitigem Abbau geschützt. Auch die Ausscheidung über die Niere wird so vermindert. Bei Verletzungen, wenn FIX benötigt wird, entweicht das Brückenprotein und rFIX kann wirken. Refixia® ist in der Europäischen Union für Patienten > 12 Jahre zugelassen.
Mit allen genannten Konzentraten können die Injektionsintervalle im Rahmen einer Prophylaxe, wie bereits erwähnt, deutlich verlängert werden (alle 1-3 Wochen) und parallel die FIX-Talspiegel zum Teil deutlich erhöht werden. Alle Präparate sind gut verträglich und wirken außerhalb der Prophylaxe auch bei der Behandlung von Blutungen oder bei operativen Eingriffen ausgezeichnet. Auch hier sind deutlich wenigere Injektionen erforderlich als unter Verwendung eines Standard-Faktor-IX-Konzentrates.
EHL-Faktorenkonzentrate in der klinischen Anwendung
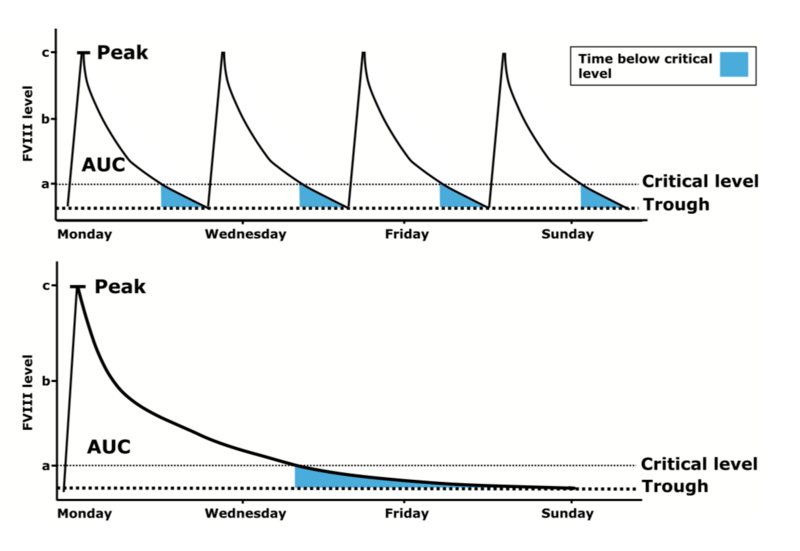
Mahdi et al, BrJ Haematol 2015 ; 169: 768-76

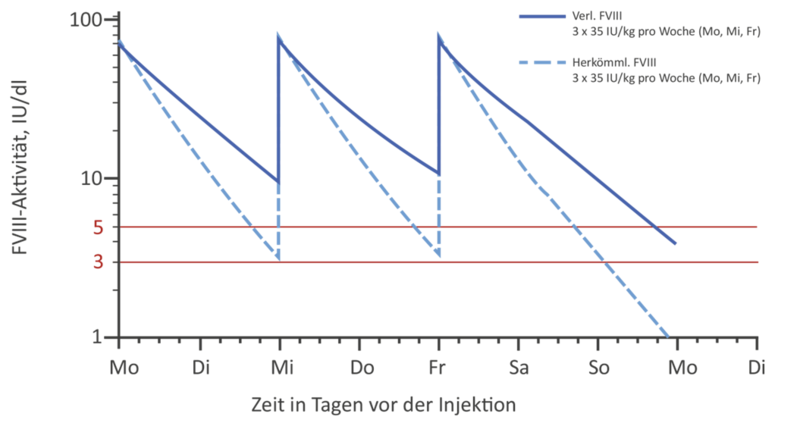
Im Vergleich zu den Standardfaktorenkonzentraten werden EHL-Konzentrate üblicherweise weniger häufig gespritzt, um den gleichen Faktortalspiegel zu erreichen. Dies trifft insbesondere auf die FIX-Konzentrate zu. Allerdings gilt zu beachten, dass zum Ende des Injektionsintervalls, insbesondere wenn sehr große Injektionsintervalle gewählt werden, nicht mehr die gewohnten, häufigen Spitzenspiegel (wie mit Standardpräparaten ca. 3 x pro Woche) erreicht werden. Wird das Injektionsintervall zu sehr in die Länge gezogen, kann es aufgrund der nun niedrigen FVIII- bzw. FIX-Spiegel zu Blutungen kommen. Dies trifft auch insbesondere dann zu, wenn der Patient zu dieser Zeit körperlich aktiv ist (s. Abb 1).
Generell bieten EHL-Faktorenkonzentrate die Möglichkeit, eine Prophylaxetherapie vielseitig zu gestalten: Soll der Talspiegel im Vergleich zum Standardkonzentrat gleich gehalten werden, kann das Injektionsintervall verlängert und damit die Prophylaxe dem Patienten erleichtert werden (s. Abb. 2a). Allerdings gibt es auch Patienten, die einen höheren Talspiegel benötigen. Hier bietet sich die Möglichkeit, bei gleichbleibenden Injektionsintervallen (z.B. bei FVIII 3 x wöchentlich) auch höhere Talspiegel zu erreichen. (s. Abb. 2b).
Unter Verwendung von EHL-Faktor-IX-Konzentraten kann aufgrund der deutlichen Halbwertszeitverlängerung zum einen eine deutliche Verlängerung der Injektionsintervalle (bis zu 3 Wochen, je nach Präparat und persönlicher Situation) bei gleichzeitigem Anheben des FIX-Talspiegels erfolgen.
Gibt es Risiken bei einer Faktorumstellung?
Üblicherweise geht eine Faktorenumstellung mit keinen nennenswerten Risiken einher. Allerdings sollte die erste Gabe im Zentrum erfolgen, um u.a. auch die Verträglichkeit zu überprüfen. Bei Patienten, die niemals einen Hemmkörper entwickelt haben, ist das Risiko, nach Umstellung einen Hemmkörper zu entwickeln, sehr gering. Allerdings sollten die Patienten ausreichend häufig vorbehandelt sein, d.h. sie sollten mindestens 150mal mit Faktorenkonzentrat behandelt worden sein. Bei Patienten, die früher einen Hemmkörper entwickelten und eine erfolgreiche Immuntoleranztherapie hinter sich gebracht haben, gibt es nur wenig Erfahrung. Dies trifft auch auf Patienten zu, die erst sehr selten mit Faktorkonzentrat behandelt wurden. Grundsätzlich sollten Sie mit Ihrem Arzt über ihr individuelles Risiko und ihr Blutungsverhalten sprechen und einen Wechsel gemeinsam abwägen.
Wie erfolgt ein Präparatewechsel? Was muss man beachten?
Zunächst sollten Sie mit Ihrem Arzt klären, ob ein Präparatewechsel für Sie sinnvoll und risikoarm ist. Eine Frage, ob und welches Präparat für Sie am ehesten geeignet ist, entscheidet Ihr Arzt aufgrund Ihrer individuellen Situation (Blutungstyp, Halbwertszeit, venöse Situation, körperliche Aktivität, Gelenkstatus, Begleiterkrankungen uvm). Besprechen Sie mit Ihrem Arzt auch Ihre Erwartungen an das neue Medikament und Ihre Erwartungen an mögliche Änderungen des Therapieregimes.
Bei Umstellung sollte die erste Gabe im Zentrum erfolgen. Sinnvoll ist es, eine pharmakokinetische Untersuchung (Halbwertszeitbestimmung) durchzuführen. Daher werden Sie anfangs u.U. zu häufigeren Visiten (Faktorspiegelbestimmung, Hemmkörperbestimmung, Fragen zur Verträglichkeit/Ansprechen auf das neue Prophylaxeregime, schnelle Erfassung von eventuellen Durchbruchsblutungen) im Zentrum gebeten. Lassen Sie sich auch über andere Rekonstitutionssysteme (wie löst man den Faktor auf?) und Lagerungsmodalitäten unterrichten. Sie sollten mit Ihrem Arzt auch Dosierungen und Häufigkeit von Faktorgaben (Dosierungsabstände) im Blutungsfall absprechen. Achten Sie auch darauf, dass Ihr Notfallausweis sowie ggf. eine Zollbescheinigung aktualisiert wird.
| Produkt | FVIII | Technologie zur Verlängerung der HWZ | HWZ-Verlängerung |
|---|---|---|---|
| Elocta® (Sobi) | rFVIII-FC, Efmoroctocog alfa | Fusionsprotein: rFVIII und Fc-Region von ImmunglobulinG (IgG) | 1,2 – 1,9 fach |
| Adynovi® (Shire) | rFVIII-PEG, Rurioctocog alfa pegol | Chemische Modifikation: PEGylierung (20kDa) von FVIII | |
| Jivi® (Bayer) | rFVIII-PEG, Damoctocog alfo pegol | Chemische Modifikation: PEGylierung (60kDa) von rFVIII | |
| Esperoct® (Novo Nordisk) | Turoctocog alfa pegol | Chemische Modifikation: Glykopegylierung (40kDa-PEG-Moleküle) | |
| Afstyla® (CSL Behring) | rFVIII, singel chain | Einzelketten-rFVIII-Molekül (Single chain rFVIII) |
| Produkt | FIX | Technologie zur Verlängerung der HWZ | HWZ- Verlängerung |
|---|---|---|---|
| Alprolix® (Sobi) | rFIX-FC | Fusionsprotein: rFIX und Fc-Region von ImmunglobulinG (IgG) | 3-5 fach |
| Idelvion® (CSL Behring) | Fusionsprotein: rAlbumin und rFIX | ||
| ReFixia® (NovoNordisk) | N9-GP | Chemische Modifikation: Ortsspezifische GlykoPEGylierung (N9GP, 40 kDa) |
Dr. Carmen Escuriola-Ettingshausen
Behandlung der Hämophilie A mit Hemlibra® (Emicizumab)
Die Hämophilie A ist eine Blutgerinnungsstörung, bei der durch einen Mangel an Gerinnungsfaktor VIII vor allem Gelenk-, Muskel- und Weichteilblutungen auftreten. Die wiederholten Blutungen führen zur Zerstörung der Gelenke und langfristig durch Arthrose zur Körperbehinderung. Daher ist eines der wichtigsten Therapieziele in der Hämophilie-Behandlung, eine Blutungsfreiheit zu erreichen. Dies gelingt in der Regel durch eine individuell angepasste Prophylaxe mit Faktor VIII-Präparaten.
In den vergangenen Jahren kamen immer wieder neue Gerinnungspräparate zur Behandlung der Hämophilie A auf den Markt. Meist handelte es sich um gentechnisch hergestellte Faktor VIII-Konzentrate, von denen vor allem die neueren Präparate durch verschiedene Verfahren, wie z.B. Pegylierung oder Fusionierung mit einem Fc-Fragment, eine verlängerte Wirkdauer aufweisen.
Mit Emicizumab wurde ein komplett neues Arzneimittel entwickelt, das sich nicht nur durch eine andere Art der Spritze („unter die Haut“ statt „in die Vene“) von den klassischen Gerinnungsfaktoren unterscheidet, sondern die komplette Therapie revolutioniert.
Was ist Emicizumab?
Emicizumab ist kein Gerinnungsfaktor, sondern ein bispezifischer Antikörper. Es handelt sich hierbei jedoch nicht um einen Faktor VIII-Antikörper, wie man fälschlicherweise annehmen könnte. Der Begriff Antikörper bezieht sich darauf, dass das Molekül Emicizumab ähnlich wie ein Schlüssel in ein Schlüsselloch passt, und statt das Signal zur „Türöffnung“ gibt Emicizumab nach Anbindung an die Gerinnungsfaktoren IXa und X das Signal zur Thrombinbildung. Das geschieht normalerweise beim Gesunden durch den ausreichend vorhandenen natürlichen Faktor VIII. Thrombin ist zur Blutstillung erforderlich.
Für welche Patienten ist Emicizumab zugelassen?
Emicizumab imitiert den Faktor VIII und ist somit bei Patienten mit einer Hämophilie A wirksam. Da es sich bei Emicizumab nicht um einen Faktor VIII handelt, kann das Medikament auch bei Patienten mit einem Faktor VIII-Hemmkörper zur Vorbeugung von Blutungen (Blutungsprophylaxe) eingesetzt werden. Das Präparat ist für Patienten mit mittelschwerer oder schwerer Hämophilie A mit oder ohne Hemmkörper zugelassen.
Was ist ein Faktor VIII-Hemmkörper?
Hämophilie A Patienten, die einen Hemmkörper (Inhibitor) gegen Faktor VIII entwickeln, können nicht mehr mit einem klassischen Faktor VIII-Präparat behandelt werden, da der Faktor VIII vom Hemmkörper zerstört wird. Um Blutungen bei den betroffenen Patienten zu behandeln oder auch um Blutungen vorzubeugen, stehen die Medikamente Feiba® (aktivierter Prothrombinkomplex) und NovoSeven® (rekombinanter Faktor VIIa) als sogenannte „Bypassing-Agents“ (BPA) zur Verfügung. Obwohl diese Therapien grundsätzlich gut wirksam sind, wird bei einigen Patienten ein Wirkungsverlust des einen oder anderen Medikamentes beobachtet. Außerdem treten trotz regelmäßiger Anwendung sogenannte Durchbruchsblutungen auf. Durch die relativ kurze Halbwertszeit werden diese Präparate zur Prophylaxe täglich oder zumindest mehrmals wöchentlich intravenös (in die Vene) verabreicht.
Wie wirksam ist Emicizumab bei Patienten mit einem Faktor VIII-Inhibitor?
In der Zulassungsstudie Haven 1 wurde die Wirksamkeit von Emicizumab bei Jugendlichen ab 12 Jahre und bei Erwachsenen untersucht, die an einer Hämophilie A mit Faktor VIII-Hemmkörpern erkrankt waren. Es konnte gezeigt werden, dass in der Patientengruppe, die vorbeugend mit Emicizumab behandelt wurde, signifikant seltener Blutungen auftraten als bei denen, die keine Prophylaxe mit Emicizumab erhielten. Patienten mit Emicizumab-Prophylaxe hatten durchschnittlich 2,9 Blutungen im Jahr. In der Gruppe, die kein Emicizumab erhielten, und die nur bei Bedarf behandelt wurden, lag die jährlich Blutungsrate bei 23,3. Nach Studiendaten ist Emicizumab zudem in der Wirksamkeit als Blutungsprophylaxe den Medikamenten Feiba® und NovoSeven® („Bypassingagents“ = BPA) überlegen. Hier konnte im Vergleich eine Blutungsreduktion um 79% erreicht werden. Patienten, die mit Emicizumab behandelt wurden, hatten in 63% der Fälle während der Beobachtungszeit sogar überhaupt keine Blutungen mehr, wohingegen Patienten ohne Prophylaxe nur in 5,6% der Fälle blutungsfrei waren. Patienten, die eine Prophylaxe mit BPA (Feiba® oder NovoSeven®) erhielten, waren in 12,5% der Fälle blutungsfrei. In der Kinderstudie Haven 2 für Patienten ab 3 Jahren wurden ebenfalls nur noch geringe Blutungsraten beobachtet. 65% der Kinder waren blutungsfrei.
In der Haven 3-Studie wurden Patienten ab 12 Jahre mit Hämophilie A behandelt, die bisher keinen Hemmkörper entwickelten. Insgesamt wurden 152 Patienten untersucht. Die jährliche Blutungsrate lag bei den Patienten, die wöchentlich mit Emicizumab behandelt wurden, bei 1,5. Patienten, die alle 2 Wochen Emicizumab in der doppelten Dosis erhielten, hatte 1,3 Blutungen pro Jahr. Patienten in der Vergleichsgruppe ohne Prophylaxe bluteten durchschnittlich 38 mal im Jahr. Diese Patienten erhielten aber auch keine Faktor VIII-Prophylaxe.
Gab es Nebenwirkungen?
In der Erwachsenenstudie Haven 1, in der Patienten mit Hämophilie A und FVIII-Hemmkörper behandelt wurden, entwickelten zwei Patienten eine Thrombose und drei Patienten eine thrombotische Mikroangiopathie (TMA). Eine TMA ist eine schwerwiegende und möglicherweise lebensbedrohliche Erkrankung, bei der die Innenwand der Blutgefäße geschädigt ist und sich Blutgerinnsel in kleinen Blutgefäßen bilden können. Dies kann zur Schädigung der Nieren und/oder anderer Organe führen. In allen diesen Fällen war zusätzlich zu Emicizumab wegen einer Blutung Feiba® gegeben worden. Bei einer genaueren Analyse der Fälle stellte sich heraus, dass eine kurzfristige zusätzliche Behandlung mit Feiba® unproblematisch war, dass aber die Gabe hoher Dosen bzw. die Behandlung über einen längeren Zeitraum zu den genannten Problemen führte. Die in den Studien behandelten Patienten entwickelten keinen Hemmkörper gegen Emicizumab.
In der Haven 3-Studie, in der Patienten ohne Faktor VIII-Hemmkörper behandelt wurden, traten keine Thrombosen und keine Hemmkörper gegen den Faktor VIII auf.
Nach Markteinführung wurde weltweit inzwischen ein Hemmkörper gegen das Medikament Emicizumab gemeldet.
Wie wird Emicizumab verabreicht?
Während bisher bekannte Gerinnungsfaktoren in eine Vene gespritzt werden, wird Emicizumab unter die Haut (subcutan) ähnlich einer Heparin- oder Insulinspritze gegeben.
Wie oft muss Emicizumab gespritzt werden?
Emicizumab wird nach einer Aufsättigungsphase mit wöchentlichen Gaben dann üblicherweise in festen Intervallen gespritzt. Das Medikament kann einmal wöchentlich, ggf. aber auch nur alle 2 Wochen oder alle 4 Wochen verabreicht werden. Welches Schema gespritzt werden soll, entscheidet der Hämophilie-Arzt.
Nach einer Aufsättigungsphase erreicht der Patient ein Dauersignal zur Blutstillung. Ein solches Dauersignal kann durch eine klassische Prophylaxe mit einem Faktor VIII-Konzentrat, bzw. mit Feiba® oder NovoSeven® nicht erreicht werden, da diese Medikamente immer wieder innerhalb weniger Stunden oder Tage abgebaut werden.
Was ist, wenn man mit Emicizumab behandelt wird und operiert werden muss?
Es gibt erste Daten zur Durchführung von Operationen mit Emicizumab: 22 von 173 Studienpatienten aus den Haven 1- und Haven 2- Studien mussten operiert werden. Bei ihnen wurden insgesamt 29 Operationen, davon 6 Zahnextraktionen, 13 Katheter-Operationen und 10 weitere OPs durchgeführt. In 7 Fällen kam es zu postoperativen Nachblutungen, von denen 3 nicht weiter behandelt werden mussten und 4 mit NovoSeven® gestillt werden konnten.
In der Haven 3-Studie wurden 50 operative Eingriffe bei 30 Patienten durchgeführt, davon 46 kleinere Eingriffe. Nur bei 18 Patienten dieser 46 Patienten wurde zusätzlich Faktor VIII gespritzt. Bei 3 Patienten kam es zu unkomplizierten Nachblutungen. Bei 4 größeren orthopädischen chirurgischen Eingriffen wurde in allen Fällen zusätzlich Faktor VIII gespritzt. Diese Operationen konnten komplikationslos durchgeführt werden.
Ist Emicizumab auch bei anderen Gerinnungsstörungen wirksam?
Emicizumab ist bei Patienten mit Hämophilie B oder anderen Gerinnungsstörungen nicht wirksam.
Welche anderen Probleme könnten unter der Therapie mit Emicizumab auftreten?
Da die Substanz die Gerinnselbildung beeinflusst, sind viele Gerinnungsteste, die die Zeit bis zur Gerinnselbildung messen, nicht mehr verwertbar. Das betrifft unter anderem die aPTT, die klassische Faktor VIII-Bestimmung im Einstufentest, die Faktor-VIII-Hemmkörper-Messung und einige andere Teste. Ein Emicizumab-Spiegel kann in Speziallaboren gemessen werden. Patienten, die mit diesem Medikament behandelt werden, müssen daher einen speziellen Ausweis erhalten, und die Therapie muss engmaschig vom Hämophiliearzt überwacht werden.
Für welche Patienten ist Emicizumab geeignet?
Emicizumab kann aktuell bei Patienten mit einer schweren Hämophilie A verschrieben werden. Dieses Arzneimittel wird eingesetzt, um Blutungen zu vermeiden oder die Anzahl der Blutungsereignisse bei Patienten mit dieser Erkrankung zu verringern. Somit ist es möglicherweise vor allem bei Patienten mit vielen Blutungen eine gute Alternative zu der bisherigen Therapie. Emicizumab wird nicht angewendet, um ein Blutungsereignis zu behandeln. In diesen Fällen muss ggf. noch ein anderes Gerinnungspräparat zusätzlich gespritzt werden.
Emicizumab ist auch nicht dazu geeignet, einen Hemmkörper zu entfernen (eliminieren). Dies ist aber für alle Patienten wichtig, damit bei ihnen zukünftig auch eine Gentherapie durchgeführt werden kann. Durch eine Gentherapie wird vom Körper „eigener Faktor VIII“ hergestellt. Dieser „eigene Faktor VIII“ würde aber durch den Hemmkörper genauso zerstört, wie der Faktor VIII aus dem Präparat, wenn es nicht gelingt, den Hemmkörper vorher durch eine Immuntoleranztherapie zu eliminieren.
Dr. Cornelia Wermes
Anti-TFPI-Therapien (Marstacimab/Hympavzi® und Concizumab/Alhemo®)
Im Jahr 2018 wurde erstmals ein Medikament zur Behandlung der Hämophilie A in Deutschland zur prophylaktischen Therapie zugelassen, das nicht mehr in die Vene gespritzt werden musste, sondern subkutan, dh. unter die Haut gespritzt werden konnte (Emicizumab/Hemlibra®). Dies bedeutete eine deutliche Erleichterung der therapeutischen Möglichkeiten, insbesondere bei Menschen mit Hämophilie A und schwierigen Venenverhältnissen. Auch Hämophilie A-Inhibitor-Patienten hatten nun eine neue Therapiemöglichkeit.
Im November 2024 erhielt dann ein ebenfalls subkutan anzuwendendes Medikament (Marstacimab/Hympavzi®) von der Firma Pfizer die Zulassung, und kurze Zeit später, Anfang 2025, folgte ein weiteres (Concizumab/Alhemo®) des Herstellers Novo Nordisk. Marstacimab ist zur Therapie bei Hämophilie A und Hämophilie B, Concizumab bei Hämophilie A und B mit Hemmkörpern in Deutschland zugelassen. Ihnen gemeinsam ist, dass sie über einen Pen (wie Sie ihn vielleicht von der Behandlung der Zuckerkrankheit kennen) subkutan unter die Haut gespritzt werden.
Es handelt sich bei beiden Präparaten um sogenannte Anti-TFPI (Tissue-Factor-Pathway-Inhibitor)-Antikörper, die die Blutgerinnung unterstützen, indem sie die Wirkung des natürlichen Antikoagulationsproteins (TFPI) blockieren. Infolge der Blockade von TFPI wird die Thrombinbildung wiederhergestellt, wodurch die Blutgerinnung gefördert wird und somit Blutungen verhindert werden. Thrombin fördert die Blutgerinnung und ist somit essenziell, um Blutungen zu verhindern. Durch die Blockierung von TFPI ermöglichen die neuen TFPI-Inhibitoren daher die Bildung von stabilen Gefäßverschlüssen und verhindern Blutungen.
Beide Präparate sollen im Kühlschrank aufbewahrt werden. Die Anwendung und die Indikationen der beiden Präparate sind unterschiedlich:
Marstacimab/Hympavzi® kann bei Patienten mit schwerer Hämophilie A und B ab 12 Jahren und einem Körpergewicht von mindestens 35 kg ohne Hemmkörper zur Prophylaxe von Blutungen eingesetzt werden. Dazu ist es notwendig einmal wöchentlich das Medikament mittels Einmal- Fertig-Pen (1 ml) subkutan zu spritzen. Die Dosierung ist initial 300 mg, danach werden 150 mg als fixe Dosis einmal wöchentlich verabreicht. Bei einem Körpergewicht von über 50 kg kann die Dosis eventuell auf 300 mg erhöht werden. Die häufigste Nebenwirkung in den Studien waren Reaktionen an der Einstichstelle (11,2%). Thrombotische Ereignisse wurden in den Studien nicht beobachtet, allerdings wurden auch keine Patienten mit vorbestehenden Thrombosen oder vermehrter Thromboseneigung untersucht.
Derzeit laufen noch Studien zur Anwendung bei Patienten mit schwerer Hämophilie A und B mit Hemmkörpern zur Prophylaxe von Blutungen sowie Studien bei Kindern. Erst wenn hier eine Zulassung erfolgt, können solche Patienten mit diesem Medikament auch behandelt werden.
Concizumab/Alhemo® kann bei Patienten mit schwerer Hämophilie A und B ohne oder mit Hemmkörpern ab einem Alter von 12 Jahren zur Prophylaxe von Blutungen eingesetzt werden. Dazu ist es notwendig, einmal pro Tag das Medikament mittels mehrfach zu verwendendem Pen subkutan zu spritzen. Es gibt verschiedene Wirkstärken (60 mg in 1,5 ml, 150 mg in 1,5 ml und 300 mg in 3 ml Lösung). Die Injektionen werden in der Regel gut vertragen. Selten gibt es Reaktionen an der Einstichstelle. Thrombotische Ereignisse wurden in den Studien nicht beobachtet. In den Studien konnte gezeigt werden, dass die Therapie auch funktioniert, wenn Hemmkörper vorhanden sind.
Achtung: Es gibt für beide Medikamente keine Indikation zur Behandlung von akuten Blutungen. Hier muss auf ein Standard-Faktor-Konzentrat ausgewichen werden.
Zusammenfassend ist mit der Zulassung der beiden neuen Medikamente eine deutliche Bereicherung in den therapeutischen Möglichkeiten bei schwerer Hämophilie A und B und bei Alhemo® bei Inhibitorpatienten erreicht worden. Weitere Studien zur Indikationserweiterung (Kinder, Inhibitoren u.A.) laufen. Derzeit müssen noch Erfahrungen mit der zusätzlichen Gabe von Faktorkonzentraten z.B. bei akuten Blutungsereignissen oder operativen Eingriffe gesammelt werden. Hier ist eine gute Rücksprache mit dem Gerinnungszentrum unbedingt anzuraten!
Dr. Günter Auerswald
Gentherapie
Update 2024
Die kürzlich in Deutschland zugelassene Gentherapie für die Hämophilie A (Roctavian) und die Hämophilie B (Hemgenix) stellt einen bedeutenden medizinischen Fortschritt dar. Bei der Gentherapie geht es darum, genetisch bedingte Krankheiten zu behandeln, indem das fehlerhafte Gen korrigiert wird. Sie ist besonders effektiv bei monogenetischen Krankheiten, also solchen, die durch einen Defekt in einem einzigen Gen verursacht werden.
Die Hämophilie eignet sich als monogenetische Erkrankung besonders gut für die Gentherapie. Durch Tansfer eines funktionierenden Gens in die Leberzelle kann eine langfristige, teilweise sogar normale Produktion des Blutgerinnungsfaktors erreicht werden. Nach einer einmaligen intravenösen Infusion des Gentherapiekonstrukts können erhöhte Faktorenspiegel erzielt werden, was selbst nach Beendigung der prophylaktischen Therapie das Risiko von Blutungen reduziert.
Neben der Gentherapie gibt es für Hämophilie bereits etablierte Behandlungsmethoden wie die prophylaktische Faktorensubstitution mit Standard- oder halbwertzeitverlängerten Faktorkonzentraten und die subkutane Gabe eines bispezifischen Antikörpers bei Hämophilie A. Diese Verfahren werden kontinuierlich verbessert, etwa durch den Einsatz noch länger wirksamer Medikamente. Daher ist es besonders wichtig, abzuwägen, welche Therapie für welchen Patienten die richtige ist. Hierbei ist zu beachten, dass die Gentherapie bei vielversprechenden Ergebnissen auch mit möglichen Risiken verbunden sein kann.
Im Folgenden soll daher ein kurzer Überblick zum gegenwärtigen Stand der Gentherapie der Hämophilie gegeben werden.
Wie funktioniert die Gentherapie?
Gentherapie ist ein innovativer therapeutischer Ansatz, bei dem Gene zur Behandlung oder Vorbeugung von Krankheiten eingesetzt werden.
Hier ist eine vereinfachte Erklärung des Prozesses:
Gen und Vektor: Bei der Gentherapie wird ein spezifisches Gen, das eine therapeutische Funktion hat (zum Beispiel ein Gen für einen Gerinnungsfaktor), in den Körper eingeführt. Dieses Gen wird zusammen mit einem Vektor transportiert. In Ihrem Fall wird ein Adeno-assoziiertes Virus (AAV) als Vektor verwendet.
Eigenschaften des Vektors (AAV): AAV sind spezielle Viren, die für die Gentherapie modifiziert wurden. Sie sind nicht-pathogen, was bedeutet, dass sie keine Krankheiten verursachen. Diese Viren können ein Gen bis zu einer Größe von 4,7 Kilobasen transportieren. Ein wichtiger Vorteil von AAV ist, dass sie nur selten ihr genetisches Material in das Genom der Wirtszelle integrieren, was das Risiko von Komplikationen wie Krebs verringert.
Kontrollelemente: Um sicherzustellen, dass das Gen im Zielorgan, hier der Leber, korrekt funktioniert, enthält der Vektor zusätzliche Elemente wie Promotoren und Enhancer. Diese Kontrollelemente helfen dabei, das Gen in die Leberzellen zu bringen und dessen Aktivität dort zu regulieren.
Synthese des Gerinnungsfaktors: Nachdem der Vektor in die Leberzellen aufgenommen wurde, beginnt die Zelle mit der Synthese des Gerinnungsfaktors, gesteuert vom eingeführten Gen. Diese Synthese findet kontinuierlich statt.
Überwachung: Nach der intravenösen Infusion des Genvektors wird die Produktion des Gerinnungsfaktors in den Leberzellen regelmäßig überwacht, um sicherzustellen, dass der Therapieansatz funktioniert und keine unerwünschten Nebenwirkungen, wie eine Erhöhung der Leberwerte, auftreten.
Für wen kommt die Gentherapie in Frage?
Die Anwendung und Auswahl der geeigneten Gentherapie für Hämophiliepatienten muss sorgfältig abgewogen und individuell auf den jeweiligen Patienten abgestimmt werden, wobei sowohl die spezifische Art der Hämophilie als auch immunologische Faktoren wie das Vorhandensein von Antikörpern gegen den Vektor zu berücksichtigen sind:
Zielgruppe: Die Gentherapie ist zugelassen für erwachsene männliche Patienten mit schwerer Hämophilie A oder schwerer und mittelschwerer Hämophilie B ohne Entwicklung eines Hemmkörpers und ohne ausgeprägte Begleiterkrankungen. Mittlerweile werden auch die ersten Studien für jugendliche Patienten mit Hämophilie geplant.
Bedeutung der Lebergesundheit: Die Leber spielt eine zentrale Rolle in dieser Therapie, da sie das Zielorgan für den genetischen Vektor und den Ort der Synthese des Gerinnungsfaktors ist. Daher ist es wichtig, dass die Leber ohne größere Funktionseinschränkung ist. Eine stattgehabte Infektion mit Hepatitis C- oder Hepatitis B-Viren stellt kein Hindernis dar, solange die Funktion der Leber nicht wesentlich beeinträchtigt ist.
Antikörper gegen AAV: Vor der Teilnahme an einer Gentherapie wird geprüft, ob Patienten Antikörper gegen Adeno-assoziierte Viren (AAV) haben. Bei bis zu 50 % der Patienten können solche Antikörper vorhanden sein. Das Vorhandensein dieser Antikörper kann die Wirksamkeit der Gentherapie beeinträchtigen, da sie die Vektoren neutralisieren könnten, bevor diese ihr Ziel erreichen.
Einfluss der Antikörper gegen AAV auf verschiedene Therapien:
Bei Hämophilie A und der Verwendung von Roctavian ist das Vorhandensein von Antikörpern gegen AAV ein Ausschlusskriterium. Bei Hämophilie B und der Verwendung von Hemgenix ist das Vorhandensein von Antikörpern gegen AAV kein automatisches Ausschlusskriterium. Es gibt Fälle, da Patienten mit nachweisbaren Antikörpern gegen AAV bereits erfolgreich behandelt wurden. Vermutlich spielt die Höhe des Antikörper-Nachweises eine Rolle.
Studienergebnisse der Gentherapie
Hämophilie B
Die ersten erfolgreichen Studienergebnisse zur Gentherapie der Hämophilie B wurden 2011 und 2014 veröffentlicht. Die Blutungsrate sank um 90%, und einige Studienteilnehmer konnten sogar ihre regelmäßigen vorbeugenden Behandlungen einstellen, ohne bei sportlichen Aktivitäten Blutungen zu erleiden. Zuletzt erfolgte ein Update zu dieser Studie auf dem ASH-Kongress im Dezember 2023, wonach über 10 Jahre und bei einem Teil der Studienteilnehmer über 12 Jahre die FIX-Werte im Wesentlichen unverändert erhöht geblieben sind. Es gab keine langfristigen Nebenwirkungen in diesem Zeitraum.
Durchbruch durch spezielle Mutation
Eine spezielle FIX Mutation wurde entdeckt, die zu einer fünf- bis zehnmal besseren Funktion des Blutgerinnungsfaktors führt. Diese Entdeckung konnte die Wirksamkeit der Gentherapie der Hämophilie B weiter verbessern.
Zwei Phase-3-Studien
Es wurden zwei Phase-3-Studien zur Gentherapie der Hämophilie B durchgeführt. Die Studie für das Gentherapieprodukt (Etranacogene dezaparvovec) führte zur Zulassung von Hemgenix. Es nahmen 54 Studienteilnehmer im Alter von 19 bis 75 Jahre daran teil. Zwei Jahre nach der Gentherapie lagen die FIX-Spiegel bei durchschnittlich 41.5 %, was selbst nach dem Stopp der zuvor bestehenden Faktortherapie zu einem Rückgang der Gelenkblutungen um 80 % führte. Das Besondere an dieser Studie war, dass auch Patienten mit dem Nachweis von Antikörpern gegen AAV behandelt wurden und in der Regel ein vergleichbar gutes Behandlungsergebnis zeigten.
Eine weitere Phase-3-Studie untersuchte das Gentherapieprodukt Fidanacogen elaparvovec bei 45 Patienten. Auch hier zeigte sich 2 Jahre nach der Gentherapie ein Ansprechen mit einer durchschnittlichen FIX-Aktivität von 25 % und einer deutlichen Reduktion der Blutungsrate im Vergleich zu dem Zeitraum vor Gentherapie.
Hämophilie A
Bei der Hämophilie A gibt es zwei wichtige Phase-3-Studien: Die Studie zu Giroctocogen Fitelparvovec, die noch Teilnehmer rekrutiert und nicht abgeschlossen ist, und die Studie zu Valoctocogen Roxaparvovec, die zur Zulassung des Medikaments Roctavian führte. In der Phase-3-Studie zu Valoctocogen Roxaparvovec erhielten Studienteilnehmer mit schwerer Hämophilie A eine einmalige Dosis des Medikaments. Bereits nach wenigen Wochen konnte ein deutlicher Anstieg der FVIII-Aktivität im Mittel um 42% erzielt werden. Im ersten Jahr nach der Infusion zeigten die Teilnehmer einen mittleren Anstieg der FVIII-Aktivität auf 23.9%. Allerdings wurde über die nächsten drei Jahre ein allmählicher Rückgang der FVIII-Expression beobachtet, mit einem mittleren Wert von 8.3% am Ende des dritten Jahres. Ein mit den Jahren stattfindender Abfall der FVIII-Aktivität wurde auch in anderen Studien zur Gentherapie der Hämophilie A beobachtet. Hierbei nahmen 17 Teilnehmer (12,7%) ihre prophylaktische Behandlung wieder auf. Trotz des Rückgangs der Faktor VIII-Expression über die Zeit hinweg zeigten die Studien klinische Vorteile während des gesamten Beobachtungszeitraums.
Welche Nebenwirkungen können nach einer Gentherapie auftreten?wurde
Nebenwirkungen können direkt während der Infusion auftreten mit allergischen Reaktionen, die medikamentös behandelt werden können.
Erhöhung von Leberenzymen: Einige Patienten weisen eine vorübergehende Erhöhung der Leberenzyme, insbesondere der Glutamat-Pyruvat-Transaminase (GPT) oder (im englischen Sprachgebrauch) Alanin-Aminotransferase (ALT) auf. Dies verläuft, ohne dass Beschwerden auftreten, kann jedoch zu einem Abfall der Aktivität der Gerinnungsfaktoren führen und somit den Erfolg der Gentherapie gefährden. Alle aufgetretenen Erhöhungen der Leberwerte konnten bisher erfolgreich mit einer vorübergehenden immunsuppressiven Therapie, wie z.B. Kortison, behandelt werden. Die Ursache hierfür ist unter anderem eine T-Zell-vermittelte Immunreaktion der Leber gegen den verwendeten Vektor.
Keine Hemmkörperbildung: Bisher sind im Rahmen der Gentherapie keine Hemmkörper gegen Faktor VIII oder Faktor IX aufgetreten.
Sehr selten Thrombosen: In sehr seltenen Fällen traten Thrombosen auf, möglicherweise bedingt durch den Einfluss deutlich erhöhter Faktorspiegel.
Theoretisches Risiko einer Tumorentwicklung: Bei vier Studienteilnehmern traten im Laufe der Jahre Krebserkrankungen auf, jedoch zeigte sich bei Untersuchungen des Tumorgewebes kein Zusammenhang mit der Gentherapie. Langfristige Datenerhebungen sind daher sinnvoll, um das theoretische Risiko einer Tumorentwicklung nach der Gentherapie zu untersuchen.
Register für Langzeitverfolgung: Für die Langzeitverfolgung der Daten zur Effektivität und Sicherheit sind Register geplant, wie z.B. das Gentherapie-Register der Gesellschaft für Thrombose- und Hämostaseforschung (GTH) und das internationale Register der World Federation of Hemophilia (WFH).
Bedeutung der Ein- und Ausschlusskriterien: Um die Nebenwirkungen der Gentherapie so gering wie möglich zu halten, ist es wichtig, die Ein- und Ausschlusskriterien genau zu beachten und eine enge Zusammenarbeit zwischen den Behandlungszentren zu pflegen.
Was ist nach der Gentherapie zu beachten?
Nach einer Gentherapie müssen Patienten verschiedene Aspekte ihrer Lebensführung beachten, um den Erfolg der Gentherapie zu sichern und mögliche Risiken zu minimieren:
Familienplanung: Es wird empfohlen, für 12 Monate nach der Gentherapie Geschlechtsverkehr nur mit Kondomen zu praktizieren und auf sichere Verhütungsmethoden zu achten, da temporär Vektorbestandteile im Sperma vorhanden sein können.
Verzicht auf Alkohol: Patienten sollten für einen Zeitraum von 6 – 12 Monaten von Alkoholkonsum absehen, um die transduzierten Leberzellen zu schützen.
Häufige Kontrolluntersuchungen: Eine sichere Teilnahme an Nachsorgeuntersuchungen ist essentiell für den Therapieerfolg. Dies kann durch die Nutzung eines Homecare Services unterstützt werden, falls notwendig.
Anforderungen an das Hämophiliezentrum
Die Anforderungen an Hämophiliezentren in Deutschland, die Gentherapien durchführen, wurden im Dezember 2023 vom Gemeinsamen Bundesausschuss (G-BA) für die Durchführung der Gentherapie Deutschland festgelegt. Diese Anforderungen zielen darauf ab, die Qualität und Sicherheit der Gentherapie für Hämophiliepatienten zu gewährleisten und eine umfassende Betreuung und Nachsorge sicherzustellen. Hierzu hat auch die GTH eine Empfehlung veröffentlicht.
Struktur- und Qualitätsvoraussetzungen: Hämophiliezentren müssen bestimmte strukturelle und qualitative Standards erfüllen, um Gentherapien anbieten zu können.
Kooperation und Koordination: Eine enge Zusammenarbeit zwischen den Hämophiliezentren ist erforderlich, insbesondere in Bezug auf Indikationsstellung, Vorbereitung und Nachbeobachtung der Gentherapie. Diese Zusammenarbeit erstreckt sich auch auf die Kooperation mit Gastroenterologen und gegebenenfalls Immunologen.
Anwendung des Hub-and-Spoke-Modell („Hub“ bedeutet „Nabe“, „spoke“ bedeutet „Speiche“): Dieses Modell, unterstützt von der Europäischen Fachgesellschaft EAHAD und der Europäischen Patientenvereinigung EHC, ermöglicht es, die verschiedenen Kenntnisse und Erfahrungen der Hämophiliezentren zu nutzen. Ein großes, spezialisiertes Hämophiliezentrum (Hub), das die Gentherapie durchführt, arbeitet eng mit kleineren Zentren (spokes) zusammen, so dass eine wohnartnahe Versorgung gegeben ist. Durch dieses Modell können Patienten auch dann behandelt werden, wenn sie weiter entfernt von den spezialisierten Zentren leben.
Darüber hinaus hat die Arbeitsgruppe Gentherapie der GTH den Einsatz elektronischer Dokumentationsmittel empfohlen. Diese sollen den Informationsaustausch zwischen allen beteiligten Parteien erleichtern und effizienter gestalten.
Zusammenfassung
Die Gentherapie für Hämophilie A und B zeigt vielversprechende Ergebnisse. Sie führt zu langanhaltenden Verbesserungen und reduziert das Blutungsrisiko nach Absetzen der Prophylaxe, was die Lebensqualität der Patienten verbessert. Bei der Gentherapie der Hämophilie B zeigen Studiendaten ein unvermindertes Ansprechen bis zu 12 Jahre nach der Therapie. Bei der Gentherapie der Hämophilie A zeigt sich ebenfalls ein gutes Ansprechen, allerdings kann die Faktoraktivität im Laufe der Zeit nachlassen.
Beim Management von möglichen Nebenwirkungen liegt ein besonderes Augenmerk auf dem Anstieg von Leberwerten, der nach der Therapie auftreten kann. Eine sorgfältige und regelmäßige Nachbeobachtung in den ersten Monaten ist daher entscheidend. Nach einer Gentherapie sind verschiedene Aspekte der Lebensführung zu beachten, um den Erfolg der Gentherapie zu sichern und mögliche Risiken zu minimieren. Hämophiliezentren, die die Therapie vorbereiten und durchführen, müssen hohe Qualitätsstandards erfüllen und eine effektive Zusammenarbeit mit anderen Zentren und anderen Fachbereichen – wie der Gastroenterologie – gewährleisten.
Da die Gentherapie noch neu ist, ist es wichtig, langfristig klinische Daten zu sammeln und in Registern zu erfassen. So können auch theoretisch mögliche Sicherheitsrisiken – wie die Tumorentwicklung – erkannt bzw. ausgeschlossen werden. Bei Kindern könnte die Gentherapie anders konzipiert werden, da ihre Leber noch wächst und sich weiter entwickelt. Hier könnten unterschiedliche Vektoren oder Technologien wie CRISPR-Cas zum Einsatz kommen, die jedoch noch in der frühen Erprobungsphase sind.
Prof. Dr. Wolfgang Miesbach, Universitätsklinikum Frankfurt
Das Europäische Hämophiliekonsortium (EHC) stellt auf seiner Homepage mehrere Informationsvideos rund um die Gentherapie bei Hämophilie zur Verfügung (auf Englisch).
Die Firma BioMarin hat eine Website mit vielen Infos über Gentherapie (allgemein sowie speziell bei Hämophilie) entwickelt.
In Zusammenarbeit mit mehreren Patientenorganisationen weltweit, auch der DHG, und der Pharmafirma BioMarin wurden zwei interessante Dokumente rund um die Gentherapie erstellt:
 .
.